Du har sannolikt någon gång köpt en blodtrycksmätare, en febertermometer eller kirurgiska ansiktsmasker i till exempel dagligvaruaffären, på apoteket eller i en webbutik. Hur vet du om produkten som du köper är adekvat? Vilket ansvar har den som säljer medicintekniska produkter?

Regleringen av medicintekniska produkter ändrades betydligt i maj 2021 i och med den nya förordningen och förändringarna som gjorts främjar produktsäkerheten. Den tidigare regleringen som varit i kraft i 25 år har i princip fungerat bra och flexibelt, men det har även funnits stora missförhållanden som nu har rättats till.
Ett av dessa missförhållanden har varit att dokumenten som intygar produktens överensstämmelse aldrig har behövt uppvisas, om inte köparen eller myndigheten särskilt begärt att få se dem.
Med den nya förordningen är det fortfarande tillverkaren som ensam ansvarar för produktens överensstämmelse, men nu har distributören ålagts nya skyldigheter, bland annat i fråga om kontrollen av produktens överensstämmelse. Vilka är de nya kraven som ställs på distributörerna och hur påverkar de försäljningen av produkterna?
Med den fria rörligheten följer även risker
CE-märkningen på produkter jämförs ofta med passet. Den garanterar produkternas fria rörlighet inom Europeiska ekonomiska samarbetsområdet. Det samma gäller även CE-märkta medicintekniska produkter.
Produkternas fria rörlighet har sin skuggsida. Det är möjligt att släppa ut på marknaden produkter vars CE-märkning beviljats på felaktiga grunder.
Produkternas fria rörlighet har sin skuggsida. Det är möjligt att släppa ut på marknaden produkter vars CE-märkning beviljats på felaktiga grunder, vilket också de facto inträffar. Sådana produkter kan komma till marknaden lika väl från Fjärran östern som från den inre marknaden inom Europeiska unionen (EU).
Tullen sköter inte marknadstillsynen av medicintekniska produkter och kan därmed inte utan grund stoppa införseln av produkter till det europeiska ekonomiska området. När man dessutom beaktar att tillverkaren har ensam ansvarat för CE-märkningen av medicintekniska produkter i den lägre riskklassen (t.ex. plåster), har det funnits attraktiva möjligheter att missbruka systemet.
Distributören måste kontrollera märkningarna på produkten
Av CE-märkningen framgår inte vilken reglering märkningen grundar sig på. För konsumenten eller säljaren av produkten har det varit svårt att ta reda på vad CE-märkningen på en produkt står för eller om den är äkta och adekvat.
I fråga om medicintekniska produkter har den nya regleringen undanröjt detta missförhållande. Utöver CE-märkningen ska det på försäljningsförpackningen anges att produkten är en medicinteknisk produkt, vilket kan göras antingen med text eller MD-symbolen (MD = medical device) (bild 1).
I medicintekniska produkter för in vitro-diagnostik har man sedan tidigare använt IVD-symbolen (IVD = in vitro diagnostic medical device) (bild 1). Det är dock bra att veta att ännu fram till våren 2025 kommer det att finnas produkter märkta enligt det tidigare regelverket som får lagligt säljas.
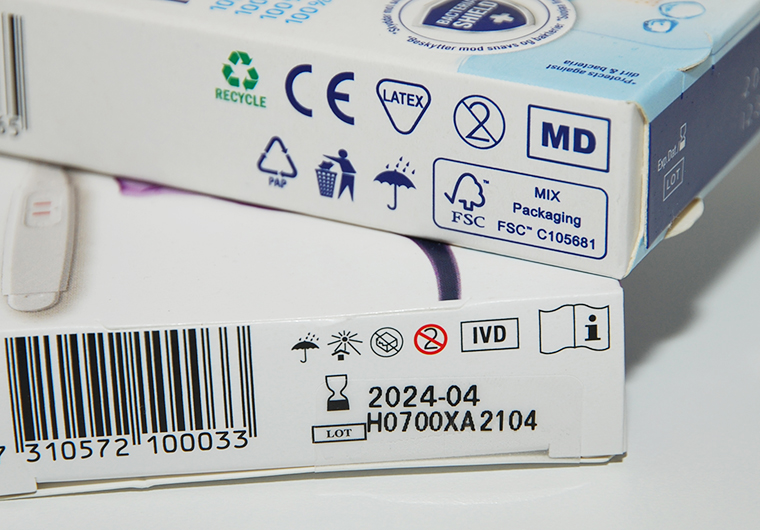
Bild 1. MD- och IVD-symboler på förpackningen av medicintekniska produkter. © Fimea
Varenda distributör i försäljningskedjan av en produkt måste säkerställa att produkttillverkaren har tagit fram de erforderliga dokumenten och märkningarna, även bruksanvisningen. Köparen i sin tur har nu bättre möjligheter att identifiera medicintekniska produkter och kan lita på att en produkt som distributören har kontrollerat är adekvat.
Effektivare tillsyn
Eventuella produkter med felaktig CE-märkning kommer också att lättare fastna i distributörernas urvalsbaserade kontroller. Om distributören har minsta lilla misstanke om att produkten inte uppfyller kraven, får distributören inte saluföra den.
Myndighetens metodarsenal har utökats i den nya nationella lagen, vilket gör det möjligt att snabbare och lättare fatta beslut om försäljningsförbud för produkter vars CE-märkning beviljats på felaktiga grunder. Tidigare försvårades utfärdandet av försäljningsförbud av att regleringen av förfarandena vid misstanke om bristande överensstämmelse med kraven inte var tillräckligt tydlig.
Utredningen av bristande överensstämmelse med tillverkaren har varit arbetsamt för myndigheten och i fråga om utländska tillverkare har man nästan alltid också varit tvungen att involvera den utländska ansvariga myndigheten i arbetet.
Registrering förbättrar spårbarheten
Det är klart att medicintekniska produkter som beviljats CE-märkning på felaktiga grunder eller som brister i fråga om egenskaperna kommer att komma till försäljning då och då även i framtiden. Nu är det möjligt att identifiera distributörerna av dessa produkter och spåra de sålda produkterna, eftersom distributörer som betjänar detaljhandlare eller social- och hälsovården måste registrera sig hos Fimea.
Dessutom måste alla distributörer kunna på begäran lämna uppgifter till Fimea om var de skaffat produkterna som de saluför och vart de har levererat produkter.
Framöver kommer identifieringen och spårningen av produkterna också underlättas av den unika produktidentifieringen (UDI) som produkterna måste förses med inom loppet av stegvisa övergångsperioder. Alla produkter måste ha denna märkning senast våren 2027. På produkten ska även information om importören som infört produkten till det europeiska ekonomiska området anges.
Ett nytt krav som avsevärt främjar produktsäkerheten är att distributörerna måste systematiskt sammanställa kundresponsen på produkterna som de levererat och rapportera den till produkttillverkaren. Om produktsäkerheten äventyras krävs också rapportering till myndigheten.
Specifikation av åtgärderna som är tillåtna för distributörerna
Den nya regleringen förtydligar och begränsar vissa åtgärder för distributörerna, till exempel ompaketering av produkter, försäljning under eget varumärke och översättning av bruksanvisningar för att uppfylla språkkraven.
Tidigare fanns inga direkt och exakta regler kring ompaketering, vilket ledde bland annat till att partiförpackningar delades upp på mindre försäljningspartier. Då kunde det hända att åtminstone delar av märkningarna och bruksanvisningarna uteblev från förpackningarna. Nu är det inte längre tillåtet att dela upp produkter som tillverkaren har förpackat till mindre försäljningsförpackningar utan adekvat bedömning av ett kontrollorgan.
Också förfarandena för översättning av bruksanvisningar har nu definierats. I förordningen anges också att distributören inte får saluföra en produkt under eget varumärke utan att komma överens om tillverkarens ansvar med den ursprungliga tillverkaren.
Också kraven på reklamföringen har utökats. Ett krav, som är synligt för konsumenten och underlättar myndighetens tillsyn, är att man i reklam måste ange att det är fråga om en medicinteknisk produkt.
Sett i sin helhet är förändringen av distributörernas ställning och skyldigheter omfattande och kräver att distributörerna på eget initiativ sätter sig in i vilka krav förordningen ställer på distributören.
Kraven i förordning (EU) 2017/745 på distributören i ett nötskal
Vi rekommenderar att man läser de exakta kraven i artikeln som anges i parentes efter texten.
En distributör är vilket som helst företag, aktör, partihandlare, butik, apotek, kiosk, person eller liknande som tillhandahåller en medicinteknisk produkt på marknaden. Tillverkaren och importören är dock inte distributörer (art. 2.34.).
Innan en produkt tillhandahålls på marknaden ska distributörerna kontrollera att
produkten är CE-märkt och att EU-försäkran om överensstämmelse avseende produkten har upprättats,
att produkten åtföljs av bruksanvisningen och de märkningar som tillverkaren ska lämna och att
informationen om importören som infört produkten i EU anges på förpackningen (art. 14).
Om distributören misstänker bristande överensstämmelse med kraven får produkten inte saluföras. Produkttillverkaren ska informeras om detta. Om distributören misstänker att produkten utgör en allvarlig risk eller är förfalskad ska också Fimea underrättas om detta. Om produkten redan har sålts ska distributören vidta åtgärder för att dra tillbaka produkten i samarbete med tillverkaren (art 14).
Distributören ska säkerställa att produktens lagrings- och transportförhållanden stämmer överens med förpackningspåskrifterna (art. 14).
Distributören ska föra register över klagomål, produkter som inte överensstämmer med kraven samt återkallelser och tillbakadraganden. Distributören ska hålla tillverkaren informerad om denna övervakning. Om distributören får klagomål eller rapporter om misstänkta tillbud med produkten som den saluför ska den omedelbart vidarebefordra denna information till tillverkaren (art. 14).
Distributören ska ta på sig tillverkarens skyldigheter om den
saluför en produkt i eget namn eller under eget registrerat varumärke utan ett avtal med tillverkaren,
ändrar det avsedda ändamålet med en produkt eller
ändrar en produkt på ett sådant sätt att det kan påverka överensstämmelsen med kraven (art. 16).
Distributören kan låta översätta den bruksanvisning som tillverkaren lämnat och lämna ytterligare information om produkten eller ändra ytterförpackningen för en produkt endast om den har ett kvalitetsledningssystem för sådan verksamhet och ett kontrollorgan har bedömt denna verksamhet. Verksamheten ska registreras hos Fimea innan den inleds (art. 16).
Distributörerna ska samarbeta med tillverkarna för att göra det möjligt att spåra produkterna. Distributörerna måste kunna lämna uppgifter till Fimea om var de skaffat produkterna som de saluför och vart de har levererat produkter (art. 25).
Förordningar
Lag om medicintekniska produkter 719/2021 (Finlex.fi)
Uppdaterad 17.12.2021 klo 16.00. Den felaktiga formuleringen gällande registrering av distributörer har korrigerats. Den rätta formuleringen är "Distributörer som betjänar detaljhandlare eller social- och hälsovården måste registrera sig hos Fimea.", inte "Detaljhandlare och distributörer som betjänar social- och hälsovården måste registrera sig hos Fimea".


.png/0fc047ef-ce40-2ea9-aa64-e1d2737bd7eb?version=1.0&t=1672664482576&width=1200)