Genom reformen av lagstiftningen vill man öka transparensen i kliniska läkemedelsprövningar och EU:s konkurrenskraft som ett område där de utförs. Hur påverkar det genomförandet av kliniska prövningar?

EU:s förordning om kliniska läkemedelsprövningar (536/2014, nedan EU-förordningen) har tillämpats från och med den 31 januari 2022. I den nationella lagen om kliniska läkemedelsprövningar (983/2021) som ska tillämpas samtidigt föreskrivs om de frågor i vilka EU-förordningen förutsätter och möjliggör nationell reglering.
Enligt EU-förordningen om kliniska prövningar av läkemedel som ska tillämpas lämnar sponsorerna in prövningsansökningar elektroniskt via EU-portalen (Clinical Trials Information System, CTIS) som upprätthålls av Europeiska läkemedelsmyndigheten (EMA) centraliserat till alla medlemsstater där man vill genomföra prövningen. CTIS-portalen är en databas som består av egna sektioner för sponsorns, de nationella läkemedelstillsynsmyndigheternas och allmänhetens bruk.
Läkemedelsmyndigheterna i medlemsstaterna bedömer ansökningarna samtidigt så att läkemedelsmyndigheten i en medlemsstat fungerar som rapporterande medlemsstat (RMS, reporting member state) medan myndigheterna i andra medlemsstater är berörda medlemsstater (MSC, member state concerned).
I EU-förordningen ingår en övergångsperiod på ett år, under vilken sponsorerna kan lämna in sin ansökan antingen enligt EU-förordningen eller enligt EU-direktivet (2001/20/EG) och enligt de utfärdade nationella bestämmelser som stöder sig på detta (Inklusive Fimeas föreskrift 8/2019). Pågående prövningar ska överföras till CTIS-portalen under övergångsperioden eller senast den 31 januari 2025. Om prövningen avslutas tidigare är det inte obligatoriskt att överföra den till portalen.
EU-förordningens bakgrund och mål
Målet med reformen har varit att öka EU:s konkurrenskraft som ett område för att genomföra kliniska prövningar. Ett harmoniserat elektroniskt bedömningsförfarande för kliniska prövningar i alla medlemsstater möjliggör att de godkänns samtidigt i flera länder med en enda ansökan. Detta innebär att alla länder har samma prövningsprotokoll (prövningsplan), vilket minskar de nationella variationerna i prövningarna.
Syftet med reformen har varit att öka EU:s konkurrenskraft som ett område för att genomföra kliniska prövningar.
Man effektiverar också samarbetet mellan medlemsstaternas myndigheter på andra sätt. Del I av utvärderingen av kliniska prövningar omfattar ett förfarande för gemensam utvärdering mellan EU-myndigheterna. Del II av utvärderingen är en utvärdering av de nationella dokumenten. Även de årliga säkerhetsöversikterna och anmälningarna om oförutsedda allvarliga biverkningar (suspected unexpected serious adverse reaction, SUSAR) bedöms på ett samordnat sätt så att det är myndigheten i en medlemsstat (safety assessing member state, SAMS) som ansvarar för den gemensamma utvärderingen av läkemedelssubstansens säkerhet.
Ett viktigt mål med EU-förordningen har också varit att öka transparensen i förfarandena för kliniska läkemedelsprövningar och informera allmänheten via den offentliga delen av CTIS-portalen. Bland annat prövningsprotokollen publiceras i portalen som standardmässiga. Syftet är att personer som är intresserade av prövningarna så enkelt som möjligt ska kunna hitta information om till exempel prövningar som gäller personens egen sjukdom. Även en lättbegriplig sammanfattning för lekmän av prövningsrapporten som publiceras efter avslutad prövning främjar medborgarnas tillgång till de nyaste prövningarnas resultat.
Prövningen delas in i två delar
Enligt bilaga I (Ansökningshandlingar för den första ansökan) i EU:s förordning om kliniska läkemedelsprövningar delas prövningen in i två delar, I och II. Sponsorn lämnar in via CTIS-systemet prövningsansökan för del I samtidigt till samtliga EU- och EES-länders myndigheter som sponsorn valt, det vill säga till läkemedelsmyndigheterna och de etiska kommittéerna. I Finland deltar både Fimea och Tukija (Nationella kommittén för medicinsk forskningsetik) i utvärderingen av båda delarna i CTIS-systemet, men samarbetar vid behov för att säkerställa att utvärderingen går smidigt till.
Del I innehåller bland annat en prövningsplan, uppgifter om de preparat som används i prövningen och prövarhandboken eller, i fråga om preparat med försäljningstillstånd, en godkänd produktresumé. Del II innehåller till exempel material som ges till försökspersonerna, uppgifter om prövarna och prövningscentren samt försäkringar. De handlingar som behövs i ansökan räknas upp i bilagorna till EU-förordningen:
- Bilaga I: Ansökningshandlingar för den första ansökan
- Bilaga II: Ansökningshandlingar för väsentlig ändring
- Bilaga III: Säkerhetsrapportering
- Bilaga IV: Innehållet i sammanfattningen av resultaten av den kliniska prövningen
- Bilaga V: Innehållet i sammanfattningen för lekman av resultaten av den kliniska prövningen
- Bilaga VI: Märkning av prövningsläkemedel och tilläggsläkemedel – en beskrivning av etiketter på prövningspreparat som används i Finland ska lämnas in med del I
I förordningens bilagor finns detaljerade anvisningar om innehållskraven för handlingarna.
Sponsorn kan göra en prövningsansökan för båda delarna samtidigt eller först för del I och inom två år för del II. Om inte ansökan för del II gjorts inom två år förfaller prövningen. Om sponsorn beslutar att ansöka först om endast del I kan ansökan om del II göras först efter att myndigheterna behandlat del I.
Behandlingstiderna från valideringen av ansökan till beslutet för del I visas på bild 1. Endast RMS kontaktar den sökande under utvärderingen av del I. Även om del I och II skulle ha lämnats in samtidigt bedöms de enligt EU-förordningen självständigt utifrån deras egna tidtabeller (bild 3).

Den som gör ansökan måste ha ett användarkonto i EMA för att ansökan ska kunna sparas i CTIS-systemet. Kontot kan ansökas från EMAs system för hantering av användarkonton (EMA Account Management) på EMA:s webbplats
Mer information om att registrera sig och detaljer om inlämnandet av ansökan finns på Fimeas webbplats (på Finska).
Validering av del I av prövningen
Före utvärderingen av prövningen validerar (förhandsgranskar) myndigheterna ansökan i enlighet med EU-förordningen (artikel 5). Sponsorn får eventuella begäran om komplettering via CTIS-systemet inom tio dagar från att prövningsansökan lämnats in.
Sponsorn ska på begäran ge kompletteringarna inom 10 dagar i CTIS-systemet. Om svarstiden överskrids förfaller prövningsansökan. Kompletteringarna bedöms inom fem dagar och om prövningsansökan konstateras vara adekvat inleder myndigheterna en bedömning av ansökan under ledning av RMS (bild 2).
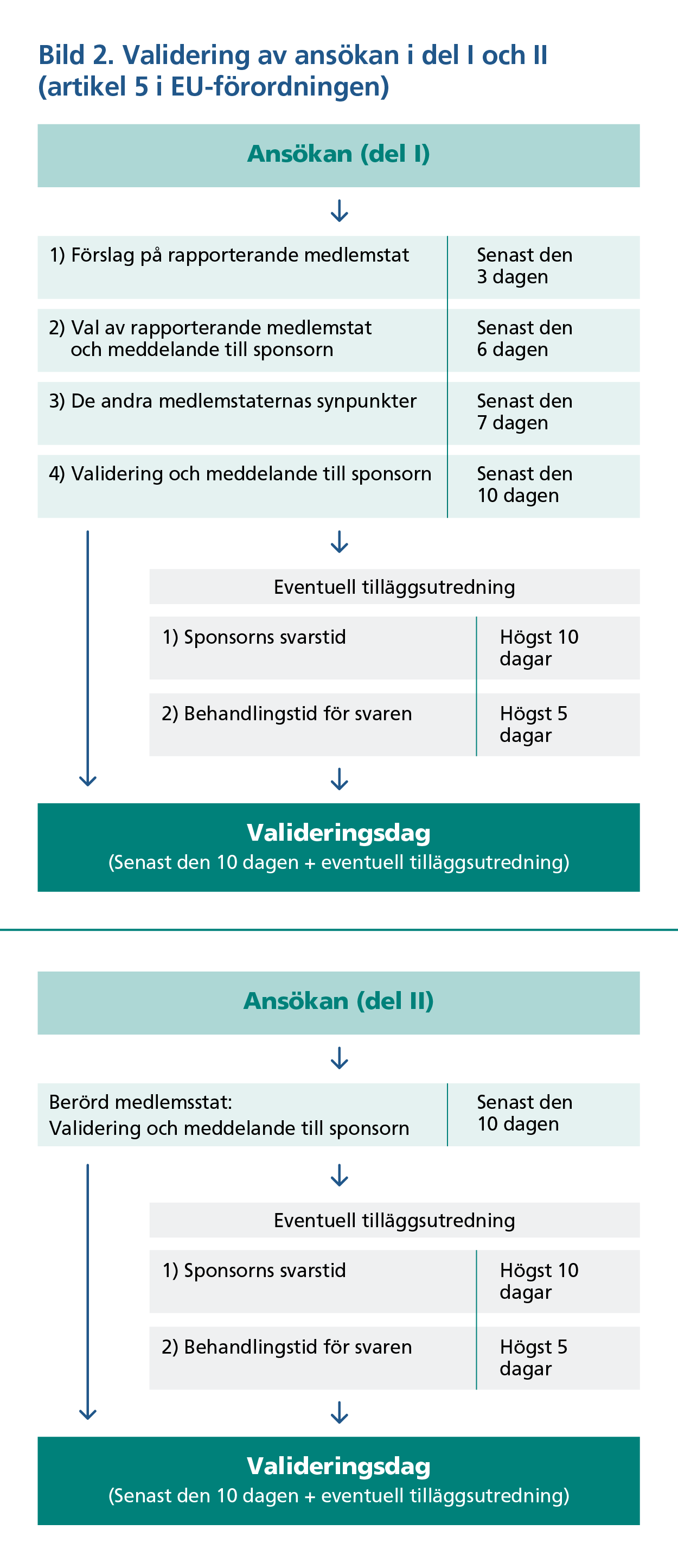
I valideringsskedet för dokumenten i del I tar Fimea ut en avgift för behandlingen, som delas med Tukija i enlighet med avgiftsförordningen.
Utvärdering av del I av prövningen
Myndighetsutvärderingen i del I av ansökan beskrivs i artikel 6 i EU-förordningen och består av tre faser: inledande utvärdering, koordinerad utvärdering och sammanställning (bild 3). RMS skall senast 26 dagar efter det att utvärderingen påbörjats utarbeta en preliminär utredningsrapport (DAR) som kommenteras och kompletteras av myndigheterna i de övriga berörda medlemsstaterna (MSC). Om det behövs kompletterande information av sponsorn skickar RMS en begäran om tilläggsutredning via CTIS-systemet.
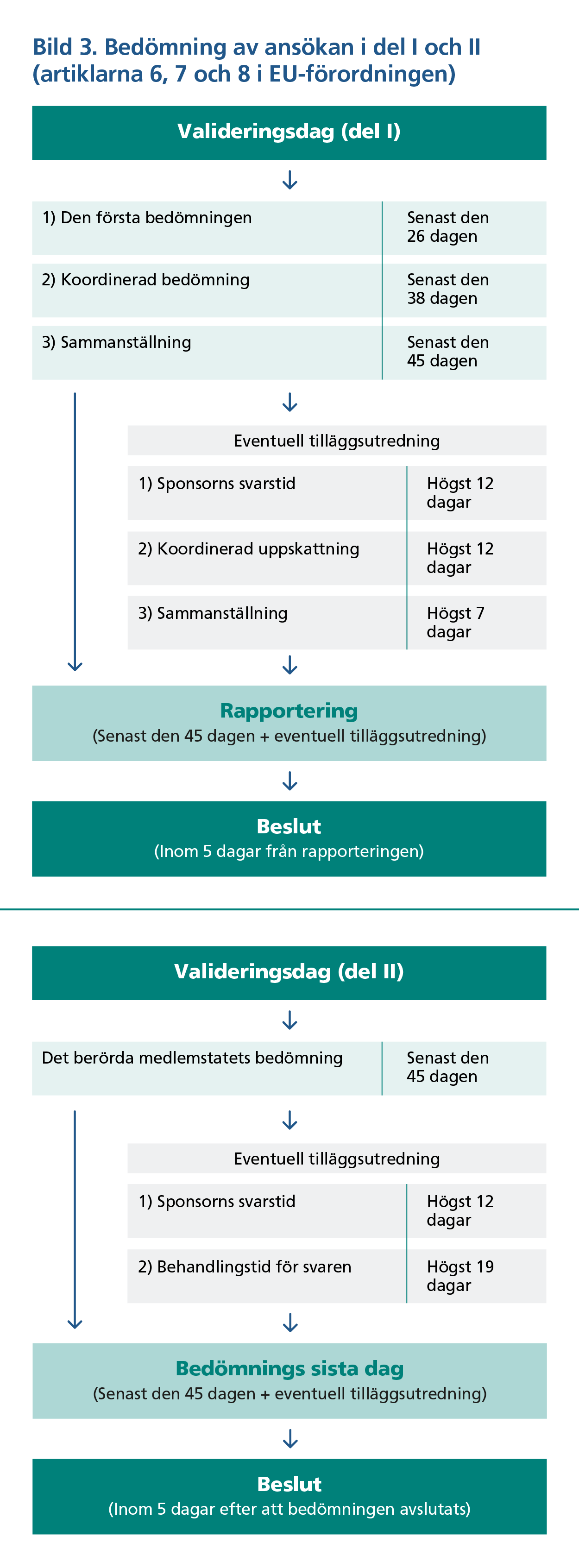
Sponsorn har en tidsfrist på 12 dagar för att lämna in den kompletterande informationen. Om svarstiden överskrids förfaller prövningsansökan. Myndigheterna utvärderar den inlämnade kompletterande informationen inom 19 dagar, varefter RMS slutliga utredningsrapport (FAR) skickas till sponsorn och de berörda medlemsstaterna (MSC) via CTIS-systemet. Utredningsrapporten ska innehålla någon av följande slutsatser om prövningen: godkänd, godkänd under vissa förutsättningar (som anges i detalj) eller inte godkänd.
Slutsatsen är bindande för alla deltagande medlemsstater. Enligt mom. 2 i artikel 8 i förordningen är det dock möjligt att ett enstaka land under vissa förutsättningar kan avvika från de andra ländernas ståndpunkt och dra en negativ slutsats, till exempel om slutsatsen bryter mot den nationella lagstiftningen.
Utvärdering av del II av prövningen
I utvärderingen av den nationella delen av prövningsansökan, det vill säga del II, deltar endast myndigheterna i landet som saken i fråga gäller. Huvudansvaret för utvärderingen av del II i Finland ligger hos Tukija. Fimea kan presentera synpunkter till Tukija för utvärderingen och utarbetandet av utredningsrapporten. Valideringens och utvärderingens förlopp i del II visas i bild 2 och 3 och dess tidsgränser följer tidsgränserna för utvärderingen i del I.
I del II finns gemensamma, av EU godkända mallar som godkänns i flera EU-länder, men man kan även använda nationella variationer. En förteckning över medlemsstaterna och de gemensamma mallarna som kan användas i varje medlemsstat har publicerats på kommissionens webbplats. På samma sida finns också andra anvisningar, bland annat ett fråge- och svarsdokument om praxis som gäller under den tid som EU-förordningen pågår.
Beslut om prövningsansökan
I Finland fattar Fimea det slutliga beslutet om prövningen inom fem dagar från att slutsatserna om både del I och del II är färdiga. Beslutet omfattar den etiska och läkemedelsmyndighetens behandling. När Fimea fattar sitt beslut är det avhängigt av en eventuell negativ ståndpunkt från Tukija.
Beslutet registreras i CTIS-systemet där det även är synligt för sponsorn. Det skickas också till sponsorn som ett elektroniskt dokument, men alla länder använder sig inte av denna praxis.
Väsentliga ändringar behandlas också i portalen
En ansökan om ändring eller tillägg av ett nytt land kan göras i CTIS-systemet när prövningen och alla tidigare ändringar har godkänts. Ett undantag utgörs av ändringsansökan för del II som kan skickas för myndighetsbehandling även om behandlingen av en ändringsansökan för del II är på hälft i ett annat land.
Valideringen av anmälan om ändring och myndighetsutvärderingen sker i CTIS-portalen ur sponsorns synvinkel på samma sätt som i den ursprungliga prövningsansökan, men behandlingstiderna för myndigheterna är något kortare (bild 4). Svarstiden för eventuell kompletterande information till valideringen (10 dagar) och för utvärderingen av ansökan om ändring (12 dagar) är densamma som för den ursprungliga ansökan.
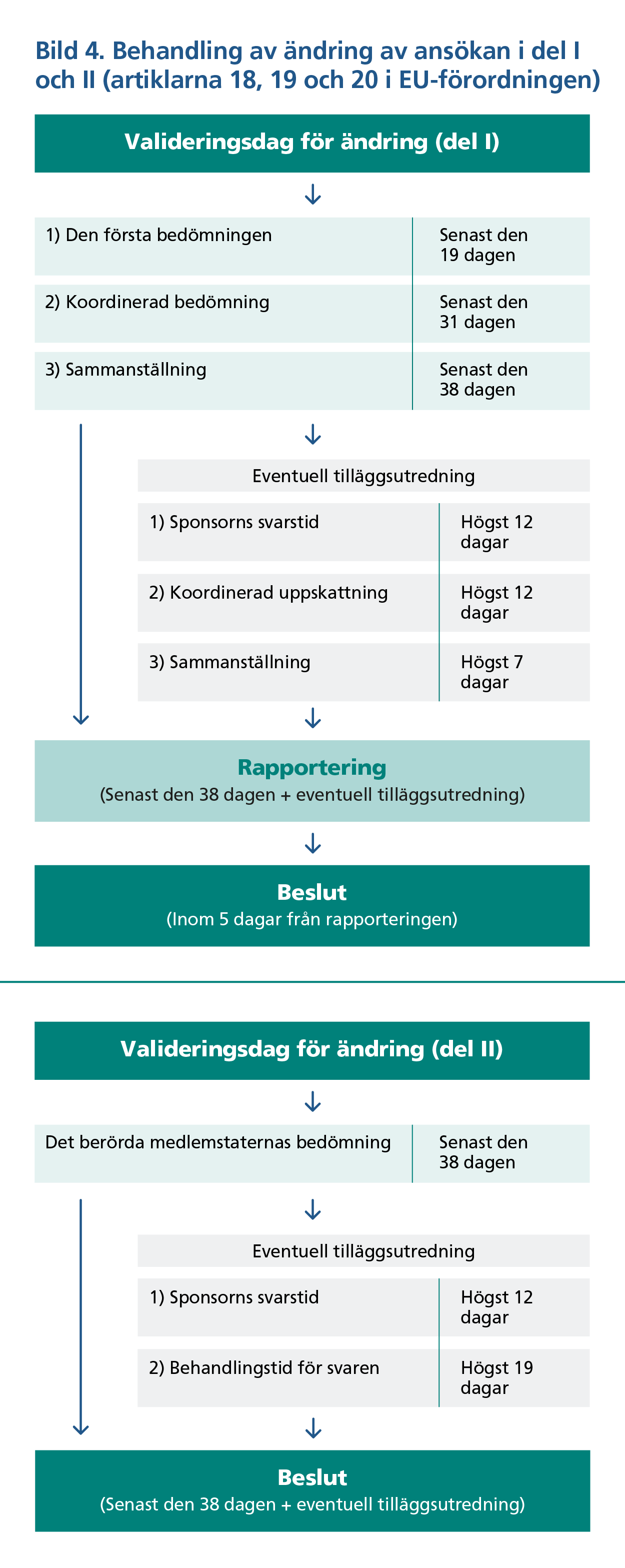
Ändringar som inte är väsentliga kan läggas till i prövningen i CTIS-systemet när inga behandlingar som gäller prövningen är på hälft.
Säkerhetsrapporteringen ska göras årligen
Sponsorn är skyldig att rapportera oförutsedda allvarliga biverkningar till databasen EudraVigilance på samma sätt som under direktivets gång. Om sponsorn, till exempel en nationell akademisk forskargrupp, på grund av brist på resurser inte kan registrera sig i EudraVigilance, kan anmälningarna om SUSAR som skett i Finland lämnas in via CIOMS-blanketten som hittas på Fimeas webbplats, varvid Fimea registrerar uppgifterna i EudraVigilance.
Sponsorn ska via CTIS-systemet årligen lämna in en säkerhetsrapport om respektive prövningsläkemedel som använts i den kliniska läkemedelsprövningen. Samma rapport kan omfatta ett eller flera prövningspreparat. Man behöver inte längre lämna in säkerhetsöversikterna nationellt.
Det bör dock observeras att om en prövning som godkänts enligt EU-direktivet genomförs från början till slut under en övergångsperiod enligt EU-direktivet, gäller fortfarande skyldigheten att lämna in en säkerhetsöversikt till Fimea. Ett undantag utgörs av prövningspreparat som också har prövningar enligt EU-förordningen och därmed även medlemsstat med ansvar för samsäkerhetsbedömningen (SaMS). SaMS utvärderar både säkerhetsrapporterna och SUSAR. SaMS skyldigheter och samsäkerhetsbedömning har föreskrivits om i EU:s genomförandeförordning om säkerhetsbedömningar av kliniska prövningar (2022/20).
Eventuella frågor om prövningens säkerhet skickas via CTIS-systemet.
Slutrapporten innehåller också en lättbegriplig sammanfattning för lekmän
Sponsorn ska spara ett sammandrag av resultaten från prövningen i CTIS-systemet senast ett år efter att prövningen avslutats (bilaga IV till EU:s förordning om kliniska läkemedelsprövningar). Resultaten av en eventuell analys under mellanperioden som är i enlighet med prövningsplanen ska också skickas till CTIS-portalen inom ett år från analysen.
Efter att prövningsansökningen godkänts blir prövningshandlingarna och myndigheternas utvärderingsrapporter offentliga och synliga i EU-databasen.
Dessutom ska sponsorn lämna in en lättbegriplig sammanfattning av resultaten i en för lekmän begriplig form (bilaga V till EU:s förordning om kliniska läkemedelsprövningar).
Syftet med EU-förordningen har varit att öka transparensen, så efter att prövningsansökningen godkänts blir prövningshandlingarna och myndigheternas utvärderingsrapporter offentliga och synliga i EU-databasen.
Undantag från offentlighetsprincipen är bland annat dokumentation om kvaliteten på prövningspreparat och myndigheternas arbetsversioner av utredningsrapporten. Sponsorn kan vid ansökan om prövning ansöka om en förlängning av offentliggörandet av prövningsdata.
CTIS-portalen vidareutvecklas
Medlemsstaternas myndigheter och Europeiska läkemedelsmyndigheten EMA följer upp de funktioner som avses i EU-förordningen och användningen av CTIS-portalen med hjälp av ett stort antal nyckeltal. CTIS-portalen vidareutvecklas och bland annat en sektion om säkerhetsutvärderingen läggs ännu till efter ibruktagandet.
Efter de eventuella första stelheterna kan man förvänta sig att särskilt utvärderingen av multinationella prövningar i Europa effektiveras och förenhetligas i och med EU-förordningen, en ökning av stöden i läkemedelsforskningsmiljön och att EU:s åtgärdsprogram för hälsobranschen i Europa genomförs.
Mer information: EU-förordningens terminologi på engelska och svenska
| engelsk term | svensk term |
|---|---|
| RMS reporting member state | rapporterande medlemsstat |
| MSC member state concerned | berörd medlemsstat |
| SaMS safety assessing member state | säkerhetsbedömande medlemsstat |
| CTIS clinical trials information system | CTIS informationssystemet och databas för kliniska prövningar |
| DAR draft assessment report | förslag till utredningsrapport |
| FAR final assessment report | slutlig utredningsrapport |
| low intervention trial | låginterventionsprövning |
| CTAG clinical trials coordination and advisory group | samordnande och rådgivande grupp för kliniska prövningar |
| serious breach | allvarlig överträdelse |
| auxiliary medicinal product | tilläggsläkemedel |
| ATMP advanced therapy medicinal product | prövningsläkemedel för avancerad terapi |
| substantial modification | väsentlig ändring |
| principal investigator | ansvarig prövare |
| incapacitated subject | försökspersonen som inte är beslutskompetent (i den nationella lagen: försöksperson som har nedsatt förmåga till självbestämmande) |
| vulnerable population | sårbara personer |
| investigator´s brochure | prövarhandbok |
| implementing regulation | genomförandeförordning |
| annual safety report | årlig säkerhetsrapport |
| urgent safety measure | brådskande säkerhetsåtgärd |
| lay summary report | lättbegriplig sammanfattning av studierapport |
Läs också
Länk till artikeln om CTIS-portalen här (på finska)
Förordningar
Lag om klinisk prövning av läkemedel 983/2021 (webbplatsen Finlex.fi)
EU:s genomförandeförordning om vid säkerhetsbedömningar av kliniska prövningar (EUR-Lex webbplats)


