EMAn hallinnoima CTIS-portaali toimii jatkossa hakemusten jättämisen, viranomaiskäsittelyn ja kaikille avoimen tiedonhaun keskuksena. Käyttöä tuetaan laajalla koulutuksella.

CTIS (Clinical Trial Information System) -portaali on Euroopan lääkeviraston EMAn hallinnoima keskitetty portaali ja tietokanta, jossa eri jäsenmaiden viranomaiset ja eettiset toimikunnat käsittelevät kliinisten lääketutkimusten hakemukset. Portaalin tarkoituksena on kliinisten tutkimusten arvioinnin ja valvonnan yhdenmukaistaminen sekä potilasturvallisuuden ja avoimuuden edistäminen EU:ssa.
Portaalissa on kaksi työtilaa – yksi viranomaisille ja toinen toimeksiantajille – sekä kaikille avoin julkinen verkkosivusto.
Toimeksiantajien työtilassa voi koota ja tehdä tutkimushakemuksia EU-alueelle samanaikaisesti yhteen tai useampaan maahan.
Viranomaisten työtilassa on hakemuksen käsittelyyn liittyviä työnkulun ja asiakirjojen hallintatoimintoja ja asiakirjojen arkistointi. Työtila mahdollistaa jäsenvaltioiden lääkeviranomaisten ja eettisten toimikuntien tutkimushakemusten yhteisarvioinnin. Euroopan komissio ja jäsenmaiden lääkeviranomaiset hyödyntävät viranomaisten työtilaa myös kliinisten lääketutkimusten valvonnassa.
EU-alueen kliinisten lääketutkimusten tiedot tulee olla julkisesti saatavilla. Tätä varten CTIS-portaalissa on julkinen verkkosivusto, josta voi etsiä tietoja hyväksytyistä kliinisistä lääketutkimuksista.
Portaali hyödyntää EMAn tietokantoja
CTIS-portaali jakaa tietoja EMAn viiden eri tietokannan ja järjestelmän kanssa. Nämä mahdollistavat käyttäjien rekisteröinnin, tiedon ja dokumenttien tallentamisen sekä organisaatioiden ja lääkevalmisteiden tietojen hakemisen CTIS-portaalista (kuva 1).
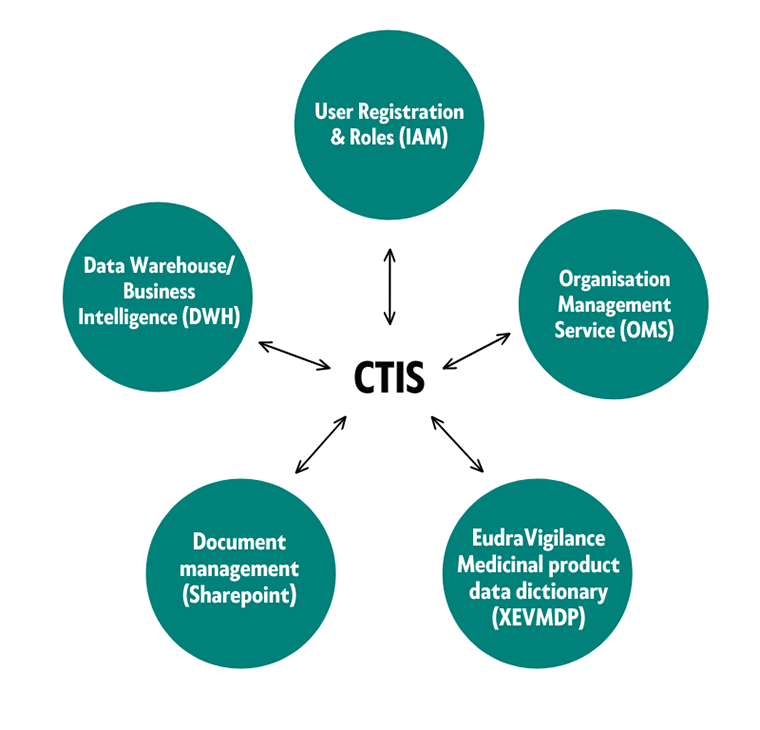
Kuva 1. CTIS-portaali on yhteydessä viiteen Euroopan lääkeviraston EMAn hallinnoimaan tietokantaan ja järjestelmään. Lähde: CTIS moduuli 2, eLearning databases and systems EMAn verkkosivuilla.
EMAn tietokannoista tulee löytyä tiedot toimeksiantajan organisaatiosta (Organisation Management Service, OMS) sekä tutkittavan- ja mahdollisen vertailuvalmisteen tiedot (EudraVigilance Medicinal Product Data Dictionary, XEVMPD) ennen tutkimushakemuksen tekemistä.
Tutkimuskeskuksen tiedot voi rekisteröidä OMS-järjestelmään hakemusta tehtäessä, ellei rekisteröintiä ole tehty aiemmin. Jos tutkimuskeskuksella on suoritettavana useita kliinisiä lääketutkimuksia, on suositeltavaa käyttää organisaatiokeskeistä rekisteröintimallia, jossa yhdellä tai useammalla käyttäjällä on käyttöoikeuksien hallinnoijan rooli. Pääkäyttäjä voi antaa muille organisaation käyttäjille erilaisia rooleja ja oikeuksia portaaliin. Toisaalta esimerkiksi yksittäisen akateemisen tutkimuksen voi rekisteröidä tietokantaan tutkimuskohtaisesti. Lisätietoa rekisteröinneistä löytyy muun muassa toimeksiantajille tarkoitetusta CTIS-pikaoppaasta.
CTIS toimii tutkimuskohtaisen tiedon säilöntäpaikkana
Tutkimuksen toimeksiantaja tallentaa hakemuksen yhteydessä CTIS-portaaliin vaaditut tiedot ja dokumentit, jotka on mainittu EU-asetuksen liitteissä. Kun tutkimus on hyväksytty, myös viranomaisen arviointiraportti julkaistaan portaalissa.
Tutkimuksen aikana toimeksiantaja kirjaa järjestelmään myös maakohtaisesti lisätietoja. Näitä ovat esimerkiksi:
- tutkittavien rekrytoinnin alku
- ensimmäisen tutkittavan ensimmäinen käynti
- tutkittavien rekrytoinnin päättyminen
- viimeisen tutkittavan viimeinen käynti
- tutkimuksen mahdollinen väliaikainen keskeyttäminen ja uudelleenkäynnistäminen.
EU-asetuksen mukaisesti toimeksiantajan tulee ilmoittaa CTIS-portaalin kautta myös vakavat sääntöjen rikkomiset (serious breach), kiireelliset turvallisuustoimet (urgent safety measures) sekä tutkittavien turvallisuuden kannalta merkitykselliset muut ilmoitukset (other reporting obligations relevant for subject safety), joista viranomainen voi tarvittaessa kysyä lisätietoja järjestelmän kautta.
Organisaatio antaa roolit omille käyttäjilleen
CTIS-portaalin käyttäjällä tulee olla EMAn käyttäjätili, jota voi hakea EMAn käyttäjätilien hallintajärjestelmästä (EMA Account Management).
CTIS-portaalissa käyttäjä voi toimeksiantajan organisaation käytäntöjen mukaisesti joko hakea oikeuksia tutkimuksen hallinnointiin tai luoda tutkimuksen itse. Kunkin organisaation tulee antaa omille käyttäjilleen roolit, joiden perusteella he pääsevät suorittamaan tutkimukseen liittyviä tehtäviä portaalissa. Ohjeita roolitukseen löytyy EMAn koulutusmoduuleista.
Huomioi siirtymäkauden aikarajat
EU-asetuksessa on siirtymäkausi, jonka aikana hakemuksia voi laittaa vireille ja niitä käsitellään joko EU-asetuksen tai EU-direktiivin (2001/20/EY) ja sen nojalla annettujen kansallisten säädösten alaisen menettelyn mukaisesti seuraavasti:
- 31.1.2022–30.01.2023: Toimeksiantaja voi jättää uuden kliinisen lääketutkimuksen hakemuksen viranomaisen käsittelyyn joko asetuksen mukaisesti CTIS-portaaliin tai direktiivin 2001/20/EY mukaisesti vain kansalliseen käsittelyyn.
- 31.01.2023 alkaen: Kaikki uudet kliinisten lääketutkimusten hakemukset käsitellään asetuksen mukaisesti eli ne tulee jättää viranomaisen arvioon CTIS-portaalissa.
- 31.01.2023–31.01.2025: Direktiivin mukaan eli kansallisesti aiemmin jätettyihin tutkimuksiin tuleva materiaali (muutos- ja päättymisilmoitukset, vuosiraportit, tulokset) käsitellään vielä direktiivin mukaisesti.
- 31.01.2025: Kaikki meneillään olevat kliiniset tutkimukset on siirrettävä CTIS-portaaliin.
Hakija voi siirtymäaikana siirtää käynnissä olevan tutkimuksensa EudraCT (European Union Drug Regulating Authorities Clinical Trials Database) -tietokannasta milloin tahansa CTIS-portaaliin. Jos tutkimus päättyy 31.1.2025 mennessä, sen siirtäminen portaaliin ei ole pakollista.
Viimeistään 31.1.2025 tutkimusten tulee olla päättyneitä tai käynnissä olevien tutkimusten siirrettyjä CTIS-portaaliin asetuksen mukaisiksi. Siirrossa tulee huomioida eettiseen arviointiin liittyvät ohjeet ja menettelyt. Tutkimustuloksia voi toimittaa EudraCT-tietokantaan myös siirtymäkauden jälkeen.
Direktiivin mukaisten tutkimusten materiaali toimitetaan Fimeaan kuten aiemmin joko CESP (Common European Submission Portal) -portaalin kautta, Eudralinkillä tai Fimean turvapostilla.
EMA ja kansalliset viranomaiset kouluttavat
Asetuksen mukaan EMAn tehtävänä on kehittää ja ylläpitää kliinisten lääketutkimusten IT-infrastruktuuria, edistää jäsenmaiden yhteistyötä muun muassa arviointiprosessissa ja tarkastuksissa sekä hoitaa turvallisuusraporttien jakelua jäsenmaille.
Vuoden 2021 alussa EMA lanseerasi sekä viranomaisille että toimeksiantajille tarkoitetun Master Trainer -koulutusohjelman, jossa EMA koulutti jokaisesta EU-maasta yhden lääkeviranomaisen ja eettisen toimikunnan edustajan CTIS-portaalin käyttöön. Lisäksi EMA on julkaissut koulutusmoduuleja portaalin toiminnasta sekä toimeksiantajille tarkoitetun käsikirjan.
EMA ja Fimea yhdessä Tukijan (Valtakunnallinen lääketieteellinen tutkimuseettinen toimikunta) kanssa ovat järjestäneet koulutusta portaalin toiminnasta, ja sitä järjestetään myös jatkossa. EMAn asiakastuki on valjastettu portaalin käyttöönotosta 31.1.2022 lähtien puolen vuoden ajan vastaamaan matalalla kynnyksellä portaaliin ja käyttäjätunnuksiin liittyvissä kysymyksissä.
Lue myös
Kliinisten lääketutkimusten suorittaminen EU-asetuksen aikana (Sic!)
Tukijan ja Fimean roolit kliinisissä lääketutkimuksissa (Sic!)
Lisätietoa CTIS-tietokannasta, käyttäjätuesta ja koulutuksesta
CTIS-käyttäjien rekisteröinti (EMA Account Management)
EMAn CTIS-asioita koskeva asiakastukipalvelu
EU-asetuksen 536/2014 ja CTIS-portaalin koulutusmoduulit (Module 23 koskee erityisesti siirtymäaikaa)
EMAn käsikirja toimeksiantajille (pdf)


