Vaikka Covid-rokotteiden kehitystyö on tehty ennätysajassa, niiden myyntilupavaatimukset ovat samat kuin minkä tahansa muunkin lääkkeen. Niiden valmistusta ja laatua arvioidaan ja valvotaan samoin kriteerein.
Normaaliin tapaan Covid-rokotteita myös tutkitaan ensin eläinkokeissa, joissa selvitetään valmisteen toksisuutta, kohde-elimiä, turvallisuusfarmakologiaa, sekä mahdollisia vaikutuksia hedelmällisyyteen ja alkion ja sikiön kehitykseen sekä poikasten kehitykseen imetyksen lopetukseen asti. Kun eläinkokeissa on osoitettu, että rokote herättää suojaavan vasteen ja merkittäviä haittoja ei havaita, voidaan siirtyä kliinisiin kokeisiin ihmisillä (kuvio 1).
Kuvio 1. Kliinisten rokotetutkimusten vaiheet eli faasit.
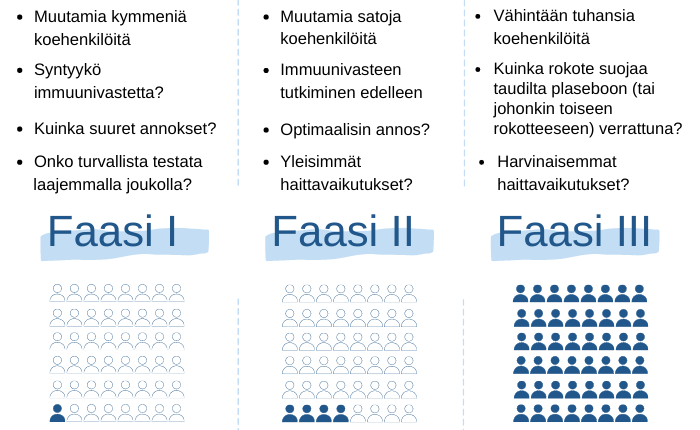
Ensimmäisessä kliinisessä vaiheessa selvitetään muutamilla kymmenillä koehenkilöillä rokotteen turvallisuutta ja muodostuvaa immuunivastetta. Toisessa kliinisessä vaiheessa varmistutaan edelleen turvallisuudesta ja testataan, synnyttääkö rokote ihmisille suojaavaa vastetta, ja valitaan myöhemmissä vaiheissa käytetty annos. Yleensä vapaaehtoisia koehenkilöitä tarvitaan muutamia satoja. Kolmannessa kliinisessä vaiheessa testataan, suojaako uusi rokote juuri siltä taudilta, jota vastaan se on kehitetty, sekä lääkkeen turvallisuutta. Usein tutkimuksessa on mukana vertailu lumelääkkeeseen. Koehenkilöitä tarvitaan yleensä tuhansia tai kymmeniä tuhansia.
EMA on julkaissut hiljattain yleisperiaatteet Covid-19-rokotteden myyntilupien vaatimuksista. Tehon osoittamiseen vaaditaan ainakin yksi hyvin suunniteltu ja laaja faasi 3 tutkimus, johon on osallistunut myös henkilöitä, joilla on jo aiemmin todettuja sairauksia, tai jotka ovat yli 65-vuotiaita - toisin sanoen myös muita kuin perusterveitä työikäisiä aikuisia.
Vähimmäisedellytyksenä on 50 %:n teho. Eli esimerkiksi 1000 ihmisestä vähintään 500:llä rokote estää koronaviruksen aiheuttaman oireisen infektion verrattuna lumekontrolliin. Turvallisuuden suhteen edellytetään riittävää rokotettavien lukumäärää ja vähintään kuuden viikon seurantaa ennen myyntiluvan myöntämistä. Tämän lisäksi vaaditaan vähintään vuoden seurantaa myyntiluvan myöntämisen jälkeen, jotta myös harvinaisemmat tai viiveellä esiintyvät haittavaikutukset tulisivat havaituiksi.
Koska täysin kattavaa tietoa lääkkeen turvallisuudesta ei normaalitilanteessakaan ole saatavilla myyntiluvan myöntämisen aikaan, myyntiluvan myöntämisen jälkeiset riskien minimointi-, hallinta- ja seurantatoimet ovat myös erottamaton osa arviointia. Niistä sovitaan myyntilupaa myönnettäessä. Myös Covid-rokotteilta edellytetään turvallisuusseurantaohjelmaa rokotteen markkinoille saattamisen jälkeen. Usein kerätään lisätietoa esimerkiksi käytöstä raskaana olevilla naisilla ja muilla erityisryhmillä.
Suhteutettuna asetettuihin vähimmäisvaatimuksiin ensimmäisten hyväksyttyjen Covid-rokotteiden (Pfizer-Biontech, Moderna) näyttö on ollut kattavaa. Kliinisissä faasin 3 ohjelmissa on ollut mukana noin 30 000–40 000 koehenkilöä, ja tehotulokset ovat olleet yli 90 prosentin luokkaa.
Keskitetty myyntilupaprosessi EU:ssa
Valtaosa Covid-19 rokotteista tullaan arvioimaan Euroopan lääkeviraston (European Medicines Agency, EMA) keskitetyn menettelyn kautta, koska tämä menettely on pakollinen kaikille bioteknologisesti valmistetuille rokotteille. Covid-rokotteiden myyntilupa-arvio on sisällöltään samanlainen kuin muidenkin lääkkeiden, mutta aikataulu on normaalia huomattavasti nopeampi ja joustavampi.
Keskitetyssä myyntilupaprosessissa EMAn lääkevalmistekomitea (CHMP, Committee for Medicinal Products for Human Use) arvioi rokotteen kehittäjien toimittamat hakemukset ja antaa suosituksen siitä, myönnetäänkö lääkkeelle myyntilupa. CHMP-komitea muodostuu yhdestä jäsenestä ja yhdestä varajäsenestä kustakin EU:n jäsenvaltiosta sekä Islannista ja Norjasta. Mukana on myös enintään viisi lisä-EU-asiantuntijaa niiltä aloilta, joilta CHMP on katsonut tarvitsevansa lisää osaamista joukkoonsa.
Kaksi toisistaan riippumatonta ryhmää arvioi
EMA nimeää uudelle myyntilupahakemukselle raportoijan ja rinnakkaisraportoijan, jotka johtavat arviointia sekä vertaisarvioijan. Ihmislääkkeiden myyntilupien raportoijina voivat toimia CHMP:n jäsenet tai varajäsenet arviointiryhmineen. Lisäksi nimetään PRAC-raportoija ja rinnakkaisraportoija, jotka vastaavat riskinhallintasuunnitelman arvioinnista. Kuhunkin tehtävään pyritään valitsemaan ne arviointiryhmät, joilla on paras asiantuntemus ja kokemus myyntilupahakemuksen aihepiiristä. Arvioijajoukkoon kuuluu farmaseuttis-biologisen laadun arvioijia, prekliinisten asioiden arvioijia, sekä kliinisten kokeiden arvioijia.
Arviointiraporteissaan kumpikin ryhmä tekee itsenäisesti yhteenvedon hakemuksen tiedoista, esittää näkemyksensä lääkkeen vaikutuksista ja mahdollisista epävarmuuksista ja lisätietoja vaativista asioista. Kaikki CHMP:n jäsenet osallistuvat aktiivisesti arviointiprosessiin kommentoimalla raportoijien tekemiä arviointeja ja esittämällä lisäkysymyksiä. Alustavasta arvioinnista ja komitean muilta jäseniltä saaduista kommenteista keskustellaan lääkevalmistekomitean täysistunnoissa. Covid-rokotteiden kohdalla hakemuksesta keskustellaan aktiivisesti prosessin aikana myös erityisessä pandemia-asiantuntijatyöryhmässä (Covid-19 EMA pandemic task force) ja muissa CHMP:n työryhmissä. Lääkekomitea päättää hakijalle esitettävistä lisäkysymyksistä.
Jos merkittäviä vastalauseita (major objections) ei voida ratkaista, ne estävät myyntiluvan myöntämisen. Nämä voivat liittyä esimerkiksi lääkkeen valmistustapaan, hyödyn kliiniseen merkitykseen, haittavaikutusten vakavuuteen, tai vaikkapa haetun käyttöaiheen laajuuteen. Arvioinnin ja hakijan toimittamien selvitysten perusteella laaditaan lopullinen CHMP:n suositus, josta päätetään virallisella äänestyksellä. Ensimmäisten covid-rokotteiden kohdalla päätös myöntää myyntilupa oli yksimielinen. Päätöksenteossa pienellä Suomella on yhtä paljon äänivaltaa kuin millä tahansa suurella EU-maalla. Euroopan Komissio päättää lopullisesti myyntiluvan myöntämisestä CHMP:n suosituksen perusteella.
Kun päätös myyntiluvan myöntämisestä tai epäämisestä on tehty, EMA julkaisee arvioinnista kattavan yhteenvedon (Euroopan julkinen arviointiraportti eli EPAR), jossa kuvataan hakemuksen tueksi toimitettu aineisto ja perustellaan, miksi CHMP suosittelee myyntiluvan myöntämistä tai epäämistä.
Myyntilupaprosessin aikataulut
Normaali keskitetty myyntilupaprosessi kestää 210 päivää, jonka lisäksi kokonaisaikaa pidentävät niin sanotut clock-stopit, joiden aikana hakijat valmistelevat vastauksia CHMP:n esittämiin lisäkysymyksiin. Jos valmiste katsotaan kriittiseksi, arviointi voidaan hakijan anomuksesta tehdä nopeutetussa aikataulussa (150 päivää).
Pandemiatilanteessa on otettu käyttöön niin sanottu ”rolling submission process”, rullaava arviointiprosessi (kuvio 2). Tämä mahdollistaa lupaavien rokotteiden myyntiluvan hyväksymisen normaalia huomattavasti nopeammalla aikataululla.
Kuvio 2. Normaalin myyntilupaprosessin ja rullaavan prosessin aikajana.
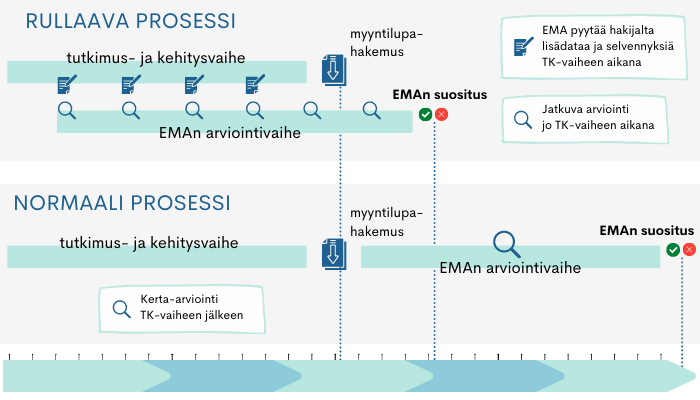
Normaalisti kaikki myyntilupahakemusta tukevat tiedot on toimitettava arviointimenettelyn alussa, mutta rullaavassa arvioinnissa tutkimustietoja arvioidaan sitä mukaa, kun niitä valmistuu. Esimerkiksi rokotteiden laatuun liittyviä ainesosatietoja ja niiden tuotantotapoja, laboratoriotutkimusten ja prekliinisten tutkimusten tuloksia on näin päästy arvioimaan jo ennen varsinaista lopullista ehdollista myyntilupahakemusta.
Koko prosessin aikataulu on hyvin joustava – hakijalta voidaan pyytää lisätietoja kesken arviointiprosessin, ja arviointisyklien (kysymys- ja arviointikierrosten) määrä ja kesto voi vaihdella. Varsinaisen myyntilupahakemuksen arviointi lopuksi tapahtuu hyvinkin nopeasti, koska suurin osa dokumentaatiosta on jo etukäteen arvioitu.
Myyntilupatyypit - tavallinen ja ehdollinen
Lääkkeelle ja rokotteelle voidaan myöntää joko tavallinen (full marketing authorisation) tai ehdollinen (conditional) myyntilupa. Valmisteen hyötyjen pitää olla aina suuremmat kuin tiedossa olevien haittojen myyntiluvan tyypistä riippumatta. Hyväksyttävät haittavaikutukset pitää aina suhteuttaa lääkkeellä hoidettavaan sairauteen ja rokotteiden kohdalla vaatimukset ovat tiukat, koska valtaosa rokotteista annetaan terveille ihmisille.
Tavallinen myyntilupa voidaan myöntää, kun hakijan toimittama dokumentaatio on niin kattava, ettei keskeisiä asioita jää selvitettäväksi myyntiluvan myöntämisen jälkeen. Ehdollinen myyntilupa voidaan myöntää vähäisemmän kliinisen näytön perusteella, jos kyseessä on lääke, jolla a) pyritään hoitamaan, ehkäisemään tai diagnosoimaan vammauttavaa tai hengenvarallista sairautta, b) jota käytetään hätätilanteessa vastaamaan kansanterveydelliseen uhkaan (kuten pandemia), tai c) jos kyseessä on harvinaislääke.
Hätätilanteessa voidaan ehdollinen myyntilupa myöntää myös silloin, kun prekliiniset ja/tai farmaseuttiset tiedot eivät ole täydelliset. Kuitenkin näissäkin tilanteissa tulee seuraavien vaatimusten aina täyttyä: tuotteen hyödyt ovat suuremmat kuin haitat; on todennäköistä, että hakija pystyy toimittamaan täydelliset tiedot sovitussa aikataulussa; puutteellisen lääketieteellisen hoidon tarpeet täyttyvät [1] (unmet medical need), ja kyseisen lääkkeen tai rokotteen välittömästä saatavuudesta markkinoilla kansanterveydelle koituvat hyödyt ovat suuremmat kuin lisätietojen puuttumiseen liittyvät riskit.
Ensimmäisten Covid-rokotteiden kohdalla kaikkien ehdollisen myyntiluvan ehtojen on katsottu täyttyneen, ja myyntiluvat on myönnetty ehdollisena. Myyntiluvan ehdoksi asetettiin erityisvelvoitteita toimittaa lisää informaatiota sekä farmaseuttisesta että kliinisestä tutkimuksesta. Näin saadaan esimerkiksi lisää tietoa pidempiaikaisesta seurannasta.
Ehdollinen myyntilupa on voimassa vuoden, ja se voidaan uusia vuosittain, jos erityisvelvoitteet on hoidettu sovitun aikataulun mukaisesti. Erityisvelvoitteiden lisäksi näihin rokotteisiin liittyy huomattavan paljon erilaisia riskien minimointi-, hallinta- ja seurantatoimia.
Fimealaisten rooli Covid-rokotteiden arvioinnissa
Suomen lääkeviranomainen Fimea ei ole toiminut Covid-rokotteiden CHMP-raportoijana. Fimea on kuitenkin osallistunut aktiivisesti kommentoivana jäsenvaltiona Covid-rokotteiden arviointiin. Fimean asiantuntijat ovat tutustuneet hakemusmateriaaleihin ja arviointilausuntoihin. Kirjallisten kommenttien ja CHMP:n ja muiden komiteoiden ja työryhmien keskustelujen kautta myös Suomen näkemykset on otettu arvioinnissa huomioon.
Suomen CHMP-jäsenen Outi Mäki-Ikolan ajatuksia prosessista
- Ensimmäisten Covid-rokotteiden keskitetty myyntilupaprosessi EU:ssa on ollut valtava ponnistus koko EU-regulaattorien verkostolle. Arviointityötä on tehty aikataulupaineen alla jokaisessa virastossa, erityisesti tietysti raportoijamaiden virastoissa.
- Eri työryhmät ja komiteat (CHMP, PRAC, BWP) ovat kokoontuneet lukuisia kertoja ylimääräisiin kokouksiin käsittelemään Covid-rokotteita (normaalisti monipäiväiset kokoukset järjestetään vain kerran kuukaudessa).
- Loppuun voisinkin silti todeta, että on ollut hienoa olla mukana tällaisessa yhteisessä ponnistelussa Fimean kollegoiden, sekä kaikkien CHMP-kollegoiden (ja PRAC, BWP, EMA) kanssa. Nämä tulevat todennäköisesti olemaan merkittävimmät päätökset, joissa kukaan meistä EU-regulaattoreista on ollut/tulee olemaan mukana työuriemme aikana.
[1] Asetus (EY) No 726/2004. Puutteellisella lääketieteellisen hoidon tarpeella tarkoitetaan sairautta, jonka diagnosoimiseksi, ehkäisemiseksi tai hoitamiseksi ei ole olemassa EU:ssa sallittua tyydyttävää tapaa ja vaikka tällainen tapa olisi olemassa, asianomaisella lääkkeellä olisi merkittäviä terapeuttisia hyötyjä sairastuneille. Lakia on tulkittu niin, että jos markkinoilla on jo täydellisellä myyntiluvalla oleva tuote saman taudin hoitoon/ preventioon, on uuden tuotteen osoitettava merkittävää terapeuttista hyötyä tähän verrattuna, jotta sille voidaan myöntää ehdollinen myyntilupa.


